Accession number là gì 2022
Thủ Thuật về Accession number là gì Mới Nhất
Quý khách đang tìm kiếm từ khóa Accession number là gì được Update vào lúc : 2022-04-01 11:57:08 . Với phương châm chia sẻ Bí kíp về trong nội dung bài viết một cách Chi Tiết Mới Nhất. Nếu sau khi đọc Post vẫn ko hiểu thì hoàn toàn có thể lại phản hồi ở cuối bài để Ad lý giải và hướng dẫn lại nha.
Bạn đang tìm kiếm từ khóa về Accession number là gì ? ví dụ code mẫu, video hướng dẫn cách sử dụng cơ bản full, link tải tải về tương hỗ setup và sữa lỗi fix full với những thông tin tìm kiếm tiên tiến và phát triển nhất được update lúc 2022-10-26 17:37:28
Nội dung chính- Video Accession number ?
- Giới thiệu BLAST trực tuyến để so sánh trình tự ADN
- So sánh cơ bản bằng công cụ trực tuyến
- Một vài lời khuyên khác
Định nghĩa Accession number là gì? Accession number là Số gia nhập. Đây là nghĩa tiếng Việt của thuật ngữ Accession number – một thuật ngữ được sử dụng trong nghành nghề nghề marketing thương mại.
:
Giải thích ý nghĩa Số tuần tự được gán cho từng hồ sơ hoặc khối lượng vì nó được thêm vào một trong những trong những cơ sở tài liệu (ví như một khuôn khổ thư viện hoặc index) và mà chỉ ra thứ tự thời hạn của việc tóm gọn về của nó. Hãy so sánh với số lượng mua. Definition – What does Accession number mean Sequential number assigned to each record or volume as it is added to a database (such as a library catalog or index) and which indicates the chronological order of its acquisition. Compare with acquisition number.
Source: Accession number là gì? Business Dictionary


Video Accession number ?
Cập nhật thêm về một số trong những trong những Review Accession number tiên tiến và phát triển nhất và rõ ràng nhất tại đây.
Chia SẻLink Download Accession number miễn phí
Quý quý khách đang tìm một số trong những trong những Chia SẻLink Download Accession number Free.
#Accession #number Nếu Quý quý khách có vướng mắc hoặc vướng mắc về Accession number thì để lại phản hồi cuối Quý quý khách nhé. Xin cám ơn đã đọc bài.
Đối với những nhà tin sinh học, tương đương (homology) là dấu vết hầu hết để Dự kiến gen và hiệu suất cao protein.
Nhưng làm thế nào để một người hoàn toàn có thể Dự kiến được xem tương đương?
Câu vấn đáp là xác lập sự tương tự giữa hai hay nhiều trình tự, tức là việc so sánh trình tự (sequence alignment).
ARN được giải trình tự, như thể những đoạn EST hay mARN khá đầy đủ hoàn toàn có thể được so sánh với trình tự hệ gen để tìm xem ở đâu có gen, đồng thời thu được thông tin về việc cải biến hoặc sửa đổi ARN.
Một ứng dụng khác nữa là phân tích SNP (những khác lạ ở một nucleotide duy nhất), khi đó trình tự ADN của mỗi thành viên được so với nhau để tìm ra những cặp base khác lạ phổ cập trong quần thể.
Có nhiều công cụ rất khác nhau để giúp ta thao tác làm ở trên, được phân loại theo thuật toán và kiểu so sánh (xem rõ ràng ở đây) nhưng những công cụ phổ cập được sử dụng để so sánh trình tự nói chung gồm có ClustalW2, T-Coffee, BLAST và FASTA3x. BLAST và FASTA vừa so sánh trình tự, vừa tìm kiếm trình tự trong CSDL. Các công cụ phải trả phí hoàn toàn có thể kể tới như DNASTAR Lasergene, Geneious, và PatternHunter.
Basic Local Alignment Search Tool (BLAST) tìm kiếm những vùng tương đương cục bộ Một trong những trình tự.
Chương trình so sánh những trình tự nucleotide hoặc protein với cơ sở tài liệu và tính toán ra mức độ trùng khớp có ý nghĩa về mặt thống kê (chứ không phải trùng khớp ngẫu nhiên).
BLAST thường được sử dụng để kết luận quan hệ về hiệu suất cao và tiến hóa Một trong những trình tự cũng như giúp xác lập những thành viên trong họ.
Giới thiệu BLAST trực tuyến để so sánh trình tự ADN
Truy cập đường link https://blast.ncbi.nlm.nih.gov/Blast.cgi, toàn bộ chúng ta có:
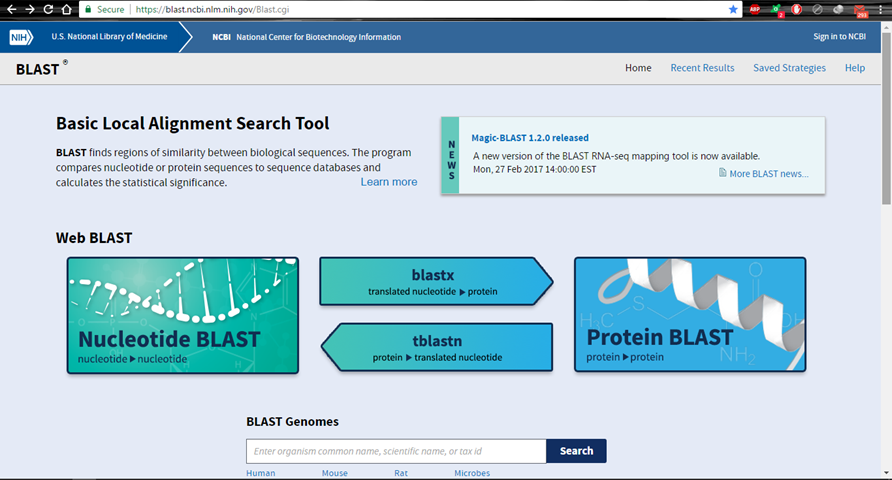
Hình 1.1. Giao diện chính của công cụ BLAST trực tuyến
Tại đây, toàn bộ chúng ta hoàn toàn có thể thấy những công cụ cơ bản:
Protein blast (blastp) để so sánh cấu trúc chuỗi amino axit cần phân tích với cấu trúc chuỗi protein trong ngân hàng nhà nước tài liệu.
Nucleotide blast (blastn): để so sánh cấu trúc chuỗi nucleotide cần phân tích với cấu trúc chuỗi nucleotide trong ngân hàng nhà nước tài liệu.
tblastn: để so sánh cấu trúc chuỗi amino axit cần phân tích với những cấu trúc protein tương ứng được dịch mã bảo toàn từ trình tự chuỗi nucleotide trong ngân hàng nhà nước tài liệu.
blastx để so sánh cấu trúc chuỗi nucleotide cần phân tích (dưới dạng được dịch khá đầy đủ sang cấu trúc chuỗi amino axit) với cấu trúc chuỗi protein trong ngân hàng nhà nước tài liệu. Phương án so sánh này được sử dụng để tìm hiểu điểm lưu ý của thành phầm sẽ tiến hành tạo ra khi lựa chọn chuỗi này.
Tiếp đó là những công cụ mở rộng và nâng cao
Hình 1.2. Một số công cụ nâng cao khác bằng BLAST (trực tuyến)
Ví dụ để so sánh trình tự ADN, chọn vào Nucleotide BLAST, giao diện của blastn như sau:
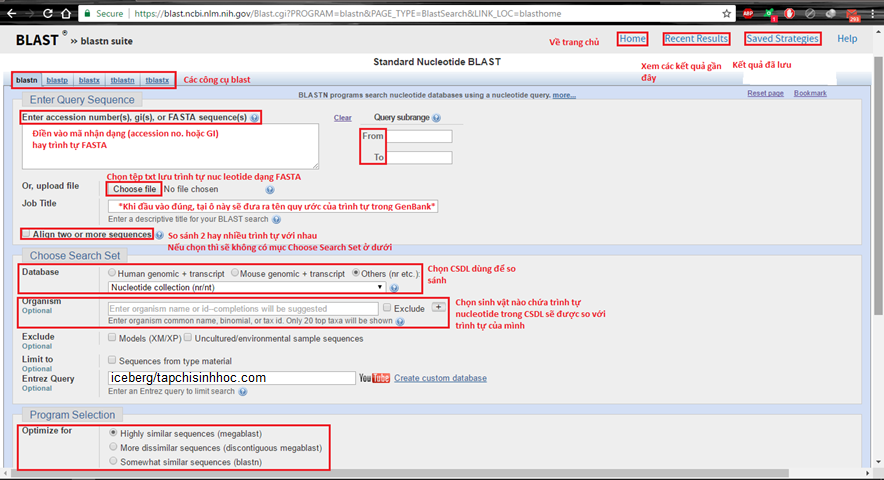
Hình 2.1 Giao diện thao tác của BLASTN trực tuyến.
Recent Results: kết quả mới gần đây được tàng trữ tự động hóa trong vòng 36h
Saved Strategies: những kết quả blast được lưu dữ thế chủ động
From – to tại Query subrange được cho phép giới hoạn một đoạn trình tự nguồn vào (Query), thay vì đưa vào toàn bộ kích thước một đoạn nào đó.
Database được cho phép lựa chọn CSDL chứa những trình tự sẽ tiến hành so sánh với trình tự nguồn vào, đó hoàn toàn có thể là hệ gene người/chuột, ARN (transcript) của người/chuột hay những CSDL khác trong số đó Nucleotide collection (nr/nt) là CSDL lớn để tạo ra phạm vi so sánh rộng.
Chọn sinh vật trong ô Organism giúp số lượng giới hạn những trình tự nucleotide trong CSDL tương ứng với đối tượng người dùng đã chọn.
Lựa chọn chính sách so sánh trình tự
Tại Program Selection, toàn bộ chúng ta hoàn toàn có thể thấy ba chính sách:
Megablast được sử dụng để so sánh truy vấn với những trình tự liên quan ngặt nghèo và tốt nhất nếu nhận dạng Phần Trăm đích là 95% trở lên nhưng rất nhanh.
Discontinuous megablast sử dụng một đoạn tương đương nhỏ ban đầu bỏ qua một số trong những base (được cho phép mismatches) và được sử dụng để so sánh Một trong những loài họ hàng; mức độ tương đương cao được kỳ vọng.
BlastN thực thi chậm, nhưng được cho phép word size hạ xuống đến 7 bases dành riêng cho việc so sánh những trình tự không thật tương đương.
Việc lựa chọn chính sách sẽ thay đổi một số trong những thông số mặc định ở phần dưới.
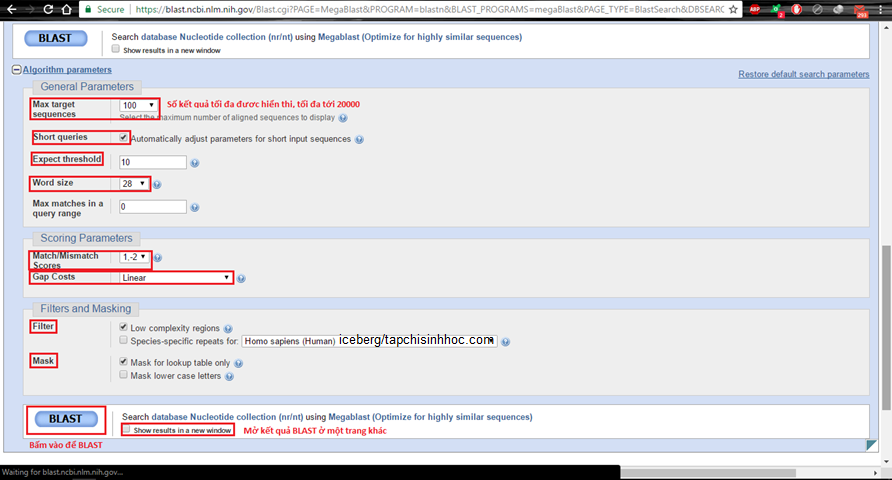
Hình 2.2. Các thông số BLAST tương ứng với chính sách megablast – Algrorithm parameters và Filters and Masking
Tại Algorithm parameters
Short Queries. Nếu ta click vào, khối mạng lưới hệ thống tự động hóa kiểm soát và điều chỉnh word size và những thông số khác để cải tổ kết quả riêng với những trình tự đưa vào ngắn.
Expect threshold. Giá trị này nêu lên ngưỡng ý nghĩa thống kê khi báo cáo những “trùng khớp” giữa trình tự truy vấn với CSDL. Giá trị mặc định threshold (T) là 10, tức là 10 bases trùng khớp được xem như không phải (hoặc ít kĩ năng) do ngẫu nhiên, theo hô hình của Karlin & Altschul (1990) (hãy nhớ rằng BLAST tìm kiếm những tương đương có ý nghĩa thống kê). Chúng ta đặt giá trị T thấp thì kết quả so sánh càng ngặt nghèo, dẫn tới ít thời cơ cho những trùng khớp ngẫu nhiên được báo cáo.
Word size: kích cỡ tối thiểu của đoạn tương đương để khởi đầu tính điểm, do đó thay đổi độ nhạy và vận tốc tìm kiếm bằng phương pháp tăng giảm word size. Theo mặc định, word size là 3 và 11 tương ứng khi so sánh protein, ADN. Trên website của NCBI được cho phép những word size rất khác nhau, hoàn toàn có thể tới 6 amino axit và 256 base. Về cơ bản, word size càng lớn thì tính ngặt nghèo càng cao, nhưng sẽ tốn nhiều thời hạn.
Filter. Các vùng trình tự có độ phức tạp thấp, tức là trình tự amino axit hay nucleotide lặp, có hàm lượng thông tin không nhiều nếu không muốn nói là rất ít, hoàn toàn có thể có ý nghĩa thống kê, nhưng không còn ý nghĩa sinh học. Ví dụ, ATATATATATATAT, PPPPPPPPPPPPPPPP hay những trình tự Alu. Chúng hoàn toàn có thể ảnh hưởng tới kết quả, nhầm lẫn, ngộ nhận, vì thế hãy ghi lại để lọc chúng tại “low complexity region filter”.
Max matches in a query range. Hữu ích khi nhiều trình tự CSDL rất khớp với một phần của trình tự đưa vào (query), điều này hoàn toàn có thể ngăn cản BLAST xuất ra những đoạn khác ít trùng khớp hơn riêng với một phần khác của trình tự đưa vào.
Match/Mismatch Scores: điểm cộng và điểm trừ tương ứng với mỗi base bắt cặp và không bắt cặp, chỉ vận dụng khi một đoạn dài hơn thế nữa giá trị T. Tỉ lệ điểm cộng/trừ nên tăng thêm khi toàn bộ chúng ta đang tìm kiếm hoặc kỳ vọng những trình tự có độ tương đương cao. Tỉ lệ 0.33 (1;-3) là thích hợp cho những trình tự bảo thủ khoảng chừng 99%; tỉ lệ 0.5 (1;-2) thích hợp hơn với những trình tự bảo thủ 95%; tỉ lệ 1 (1;-1) tốt nhất cho bảo thủ 75%. Điều này nghĩa là, nếu toàn bộ chúng ta đặt Match/Mismatch Scores là (1;-1) thì kết quả xuất ra sẽ gồm có cả những trình tự có độ tương đương khoảng chừng 75% trở lên. Ở chính sách Megablast, giá trị này mặc định là (1;-2).
So sánh cơ bản bằng công cụ trực tuyến
So sánh một trình tự với cả CSDLMuốn so sánh phải có Query – trình tự nguồn vào – là trình tự mà toàn bộ chúng ta đang sở hữu/đang quan tâm và muốn so sánh với trình tự khác. Các phương pháp để lấy trình tự vào BLAST?
Hãy khởi đầu với việc tìm kiếm gene quan tâm trên CSDL Nucleotide của NCBI. Sao chép Accession hoặc GI ngay tại trang kết quả trả về.
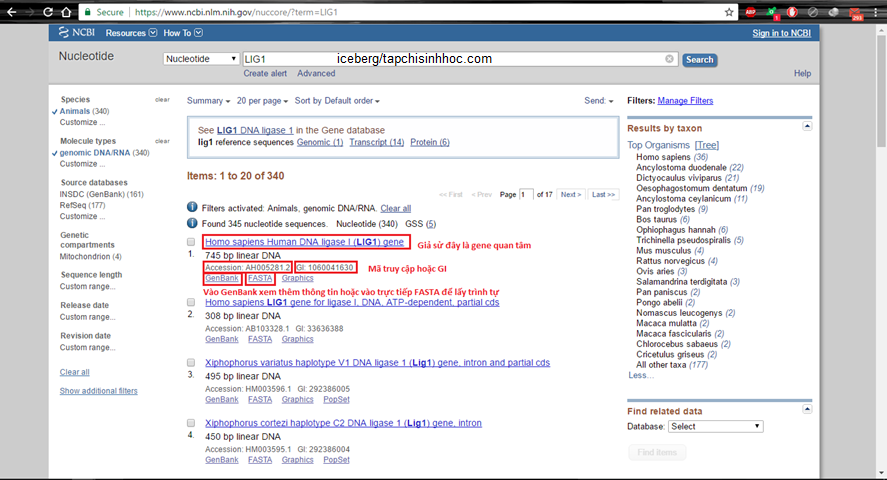
Hình 3.1. Tìm kiếm mã truy vấn của gene ở CSDL nucleotide
Cách 1. Dùng mã truy vấn Accession number hoặc GI
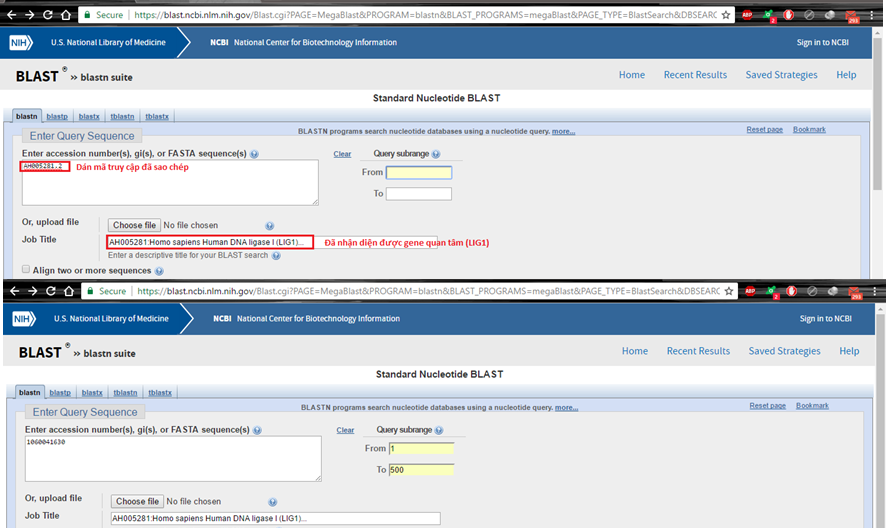
Hình 3.2. Nhập vào trình tự Query bằng Accession number hoặc mã GI
Trong trường hợp này, toàn bộ chúng ta cần nêu lên số lượng giới hạn của đoạn trình tự mà ta quan tâm mà thôi, tại From và To
Cách 2. Vào FASTA lấy trình tự
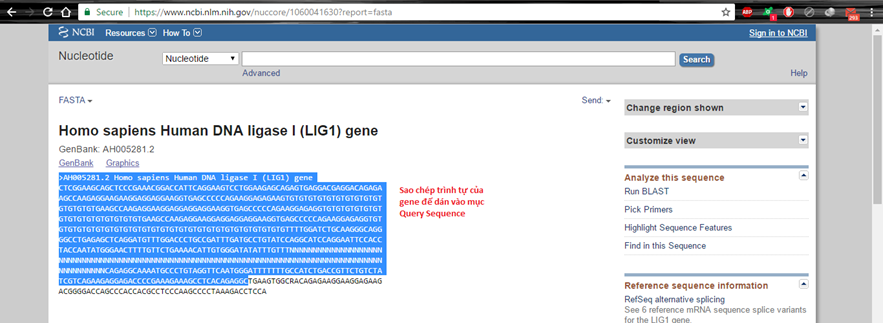
Hình 3.3. Truy cập FASTA Nucleotide để lấy trình tự gene
Hãy dán trình tự đã sao chép vào Query
Xem thêm: cách tìm kiếm gene và lấy trình tự trên NCBI
Cách 3. Tải lên tệp tin text
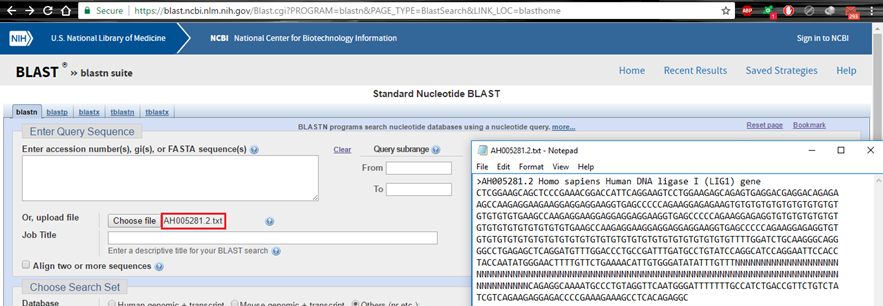
Hình 3.4. Nhập vào trình tự Query bằng tệp tin txt có sẵn
Lưu ý, file khả dụng có đuôi .txt hoặc .fasta; ngoài ra, dòng thứ nhất trong file nên phải có dòng định danh gene , tối thiểu là phải có một ký tự ‘>’ (“>Accession number …”).
Sau khi đã nhập, chọn chính sách blast là megablast, chọn mở kết quả sang tab mới, bấm BLAST. Khi kết quả BLAST lần đầu hiện ra, toàn bộ chúng ta hoàn toàn có thể vẫn cần sửa đổi một số trong những thông số khác, vì thế mở kết quả sang tab mới để tránh làm mất đi kết quả lần thứ nhất.
Hình 4. Màn hình chờ khi đang Blast
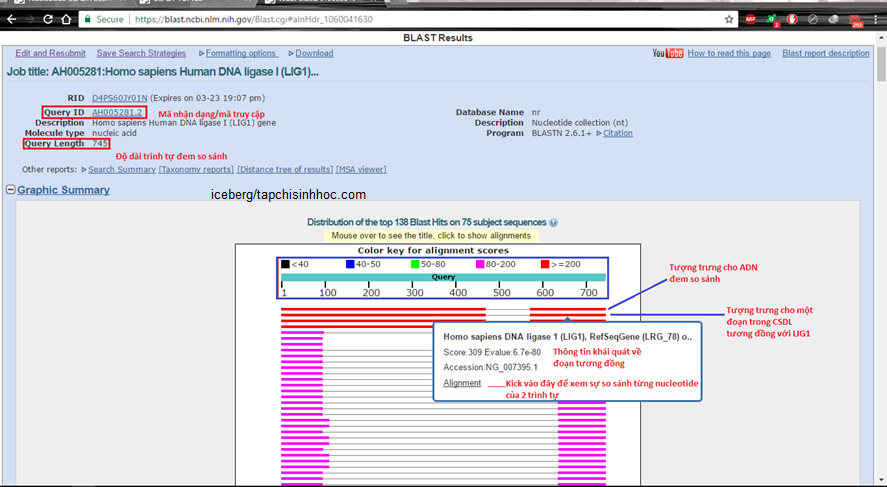
Hình 5.1. Phần đầu kết quả sau khi BLAST xong
Phần phía trên thể hiện những thông tin về trình tự nhập vào, gồm có ID và độ dài. Lưu ý là kết quả so sánh này chỉ tồn tại trong thuở nào gian (36 h), nên nếu bạn có lưu trang lại mà tiếp theo đó không hề kết quả thì cũng dễ hiểu.
Graphic Summary. Thang màu thể hiện mức độ tương đương của một trình tự trong CSDL và trình tự đem so sánh (query): màu đen thể hiện số bases tương đương dưới 40, tăng dần đến trên 200 bases tương đương thể hiện bằng red color.
Khi trỏ vào mỗi trình tự từ thứ hai trở đi sẽ là thông tin về trình tự trong CSDL tương đương với trình tự đem BLAST; nhấn vào Aligment sẽ dẫn tới phần so sánh trình tự nu của 2 mạch (đề cập sau)
Các kết quả được liệt kê theo thứ tự giảm dần về sự việc tương đương.
Descriptions: xem những mô tả về toàn bộ những trình tự có mức độ tương đương.
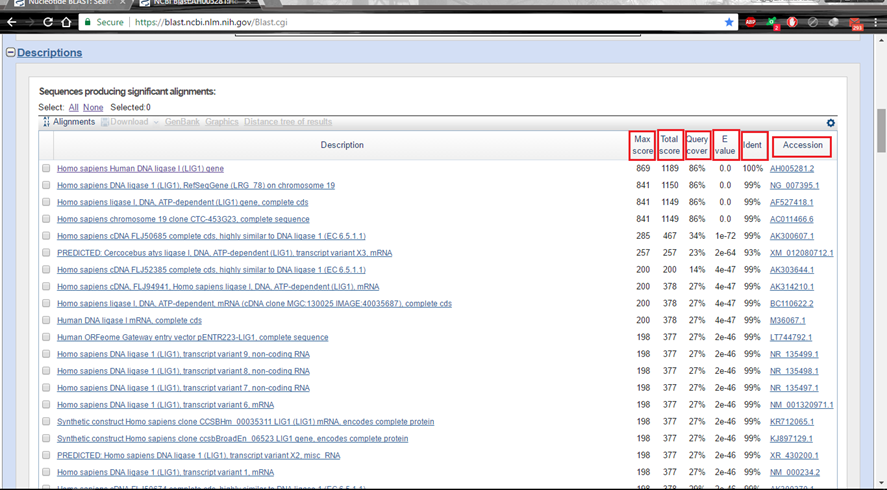
Hình 5.2. Mô tả những thông số BLAST của từng bản ghi
Max score là yếu tố cao nhất lúc BLAST trình tự truy vấn với CSDL, ứng với đoạn tương đương nhất (dài nhất) giữa 2 trình tự, góp thêm phần rất rộng vào Total score.
Total Score là tổng điểm khi BLAST, sinh ra từ những đoạn tương đương giữa 2 trình tự. Với mỗi vùng tương đương (to nhiều hơn hoặc bằng word size) thì BLAST khởi đầu tính điểm. Nếu Total score bằng Max score thì tức là chỉ có duy nhất một đoạn tương đương; còn nếu Tatal score to nhiều hơn Max score thì tức là có nhiều đoạn tương đương giữa 2 trình tự và Total score bằng tổng điểm của những đoạn ấy.
Query cover là tỉ lệ bao trùm (tính qua độ dài) của trình tự đưa vào so với trình tự tương đương tìm thấy (hit) trong CSDL. Nếu dài bằng nhau tức là bao trùm 100%.
E value (liên quan tới Expect threshold) là mức độ giống nhau ngẫu nhiên của những đoạn tương đương giữa 2 trình tự. E value trong BLAST sẽ tính đến hơn cả chiều dài và thành phần đoạn tương đương cùng với tỉ lệ Phần Trăm tương đương. E value càng nhỏ tức là những hit tìm thấy càng có ý nghĩa thống kê – tính ngẫu nhiên là càng thấp. Cùng một trình tự nhưng tìm kiếm trên những CSDL rất khác nhau sẽ cho E rất khác nhau, là bởi số trình tự có sẵn (tham chiếu) trong CSDL. Tóm lại, E-value là một chỉ số đại diện thay mặt thay mặt cho ý nghĩa thống kê.
Identity là mức độ tương đương giữa 2 trình tự – kết quả toàn bộ chúng ta mong đợi nhất lúc thực thi phép so sánh trình tự.
Trong nhiều trường hợp, toàn bộ chúng ta nên quan tâm nhất đến tỉ lệ bao trùm và mức độ tương đương. Hai trình tự phải không thật ngắn, có mức độ bao trùm càng nhiều và mức tương đương càng cao thì sẽ càng chỉ ra tính giống nhau giữa chúng.
Tiếp theo, nhờ vào những bản ghi để xem gene đang so sánh hoàn toàn có thể là gene gì hoặc có vai trò gì. Bấm vào mỗi bản ghi để thấy hiệu suất cao. Điều này là hữu ích khi toàn bộ chúng ta vừa giải trình tự một gene: toàn bộ chúng ta được gợi ý được vai trò của gene đó hoặc kiểm tra xem gene đó có đúng là gene mong ước.
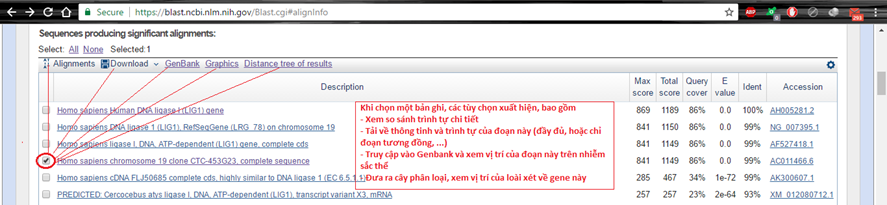
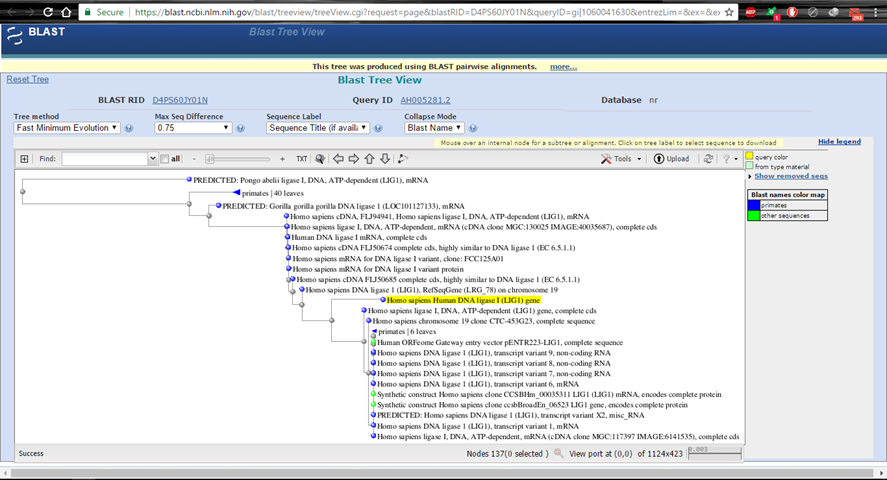
Hình 5.3. BLAST tree view
Cuối cùng tại Aligment, xem so sánh rõ ràng trình tự nucleotide Query với những hit tìm thấy. Tại đây toàn bộ chúng ta thấy được những khác lạ nhỏ giữa hai trình tự, như mismatch, gap và vị trí tương đương.
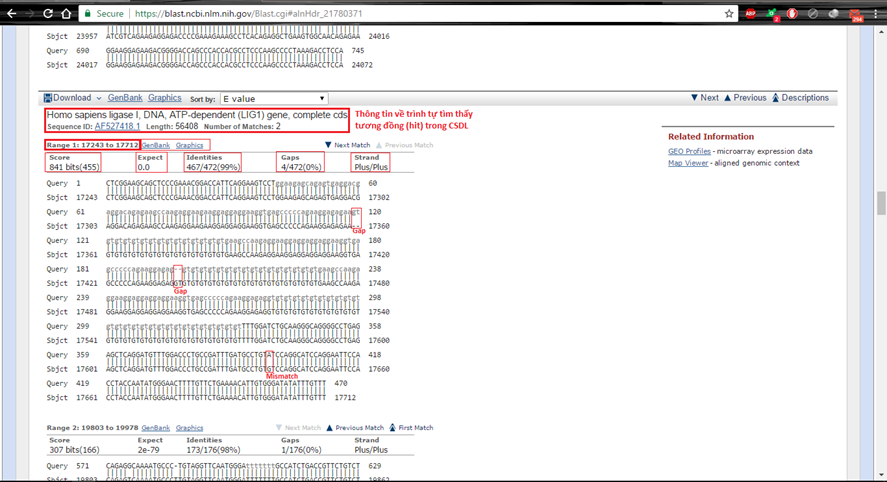
Hình 5.4. So sánh Query với một hit dưới dạng trình tự nucleotide
So sánh gene tương đương trên hai sinh vật bằng BLASTĐầu tiên, toàn bộ chúng ta chỉ việc chọn Align two or more sequences. Phần tiếp theo cũng rất tương tự như trên, ta có 3 phương pháp để lấy trình tự vào so sánh. Việc lựa chọn CSDL là không thiết yếu nữa, tuy nhiên toàn bộ chúng ta vẫn hoàn toàn có thể tùy chỉnh những thông số BLAST phía dưới. Kết quả so sánh 2 trình tự:
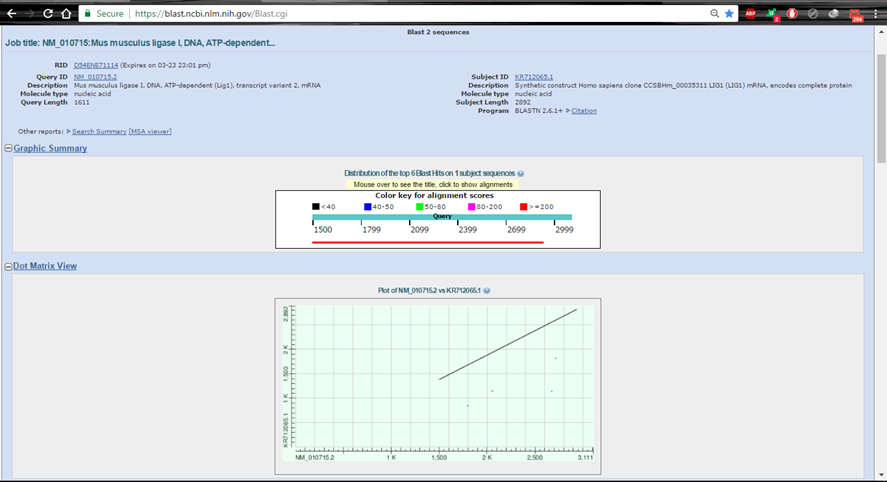
Hình 6. Graphic Summary và Dot matrix (ma trận tính điểm) khi BLAST 2 trình tự mRNA
Một vài lời khuyên khác
Đừng bất thần và hãy nhớ rằng toàn bộ chúng ta hoàn toàn có thể không thu được cùng một kết quả khi bạn chạy blast (cùng những thông số đó) ở những thơi điểm rất khác nhau.
Các bản update riêng với CSDL hoàn toàn có thể làm thay đổi kết quả so sánh, ví dụ CSDL ban đầu chỉ có 100 trình tự thì giờ đây số lượng là 250, nên nhiều kĩ năng số kết quả sau blast hoàn toàn có thể sẽ phong phú hơn.Tốt hơn là cứ thiết đặc những thông số ở dạng mặc định, chỉ khi điều này không khả thi, hãy sử dụng những gì bạn đọc được ở đây, biết đâu lại tìm ra một kết quả thích hợp hơn.Ngoài ra, cũng không còn mức giá trị “thần thánh” nào xác lập kết quả mà CSDL trả cho bạn thực sự khớp với trình tự truy vấn của bạn. Để là một người tiêu dùng linh hoạt, hãy học cách tùy chỉnh những tham số mà chúng tôi thảo luận ở trên.
Nguồn tìm hiểu thêm chính: NCBI BLAST help page.
iceberg (tổng hợp và trình diễn)
tapchisinhhoc.com
Share Link Download Accession number là gì miễn phí
Bạn vừa tìm hiểu thêm nội dung bài viết Với Một số hướng dẫn một cách rõ ràng hơn về Video Accession number là gì tiên tiến và phát triển nhất và Chia Sẻ Link Down Accession number là gì miễn phí.